Mato Grosso
Bebê apela à Justiça para ter acesso ao medicamente mais caro do mundo
Com uma doença degenerativa, criança precisa receber o tratamento nos próximos 5 meses
Saúde | 18 de Outubro de 2024 as 12h 41min
Fonte: Redação com assessoria

No próximo dia 12 de novembro, o pequeno Caê Nogueira será submetido a uma perícia médica, agendada pela Justiça Federal de Sinop, no município de Sorriso. A avaliação do médico vai ajudar a determinar se o bebê de um ano e 7 meses terá acesso ao Zolgensma, o mais promissor medicamento para o tratamento de AME (Atrofia Muscular Espinhal), uma doença degenerativa e genética que atinge os neurônios da medula espinhal, interferindo na capacidade de andar, mover, engolir e até respirar, podendo levar à morte.
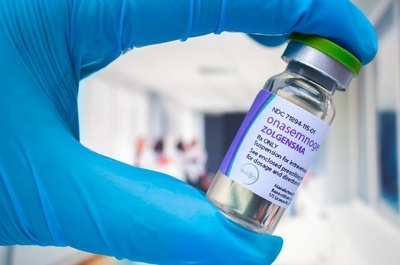
O apelo à Justiça é para que o SUS (Sistema Único de Saúde), forneça o Zolgensma para Caê. O medicamento, conhecido como o “mais caro do mundo”, custa cerca de R$ 7,5 milhões. O tratamento, além de caro e raro também é limitado pelo tempo. A dose única do Zolgensma precisa ser aplicada antes da criança completar 2 anos de idade. Ou seja, Caê precisa vencer essa batalha na Justiça ante do dia 8 de março de 2025.
A ação judicial corre pela 21ª vara da Justiça Federal, em Brasília. Como a família de Cauê reside em Lucas do Rio Verde (MT), foi expedida uma carta precatória para a Justiça Federal de Sinop, afim de que a perícia médica fosse realizada mais perto do domicílio. A Carta Precatória foi emitida no dia 23 de setembro e no dia 2 de outubro o juiz federal de Sinop, André Perico dos Santos nomeou o médico perito Samuel Alves para realizar o procedimento. Ele tem 30 dias para apresentar o laudo que dirá se o pequeno Caê deve ou não ter acesso ao Zolgensma pelo SUS.
A história de Caê
Depois de anos juntos, Ewerton Costadelle e Larissa Nogueira decidiram que era hora de ter um filho. A gravidez planejada, devidamente acompanhada, com uma série de exames médicos, guardou uma surpresa: Larissa esperava gêmeos.
A gestação transcorreu de forma normal, sem complicações. Com um parto cesariana na 37ª semana, vieram ao mundo Tom e Caê. Tom era um pouco maior que seu irmão, mas até os 6 meses de vida, ambos se desenvolveram normalmente.
Com o passar do tempo, Larissa e Ewerton começaram a perceber diferenças entre o crescimento de Caê e Tom. Enquanto Tom já demonstrava uma certa mobilidade, Caê só conseguiu se sentar aos 8 meses de idade e engatinhou só aos 10 meses – tempo que está dentro dos marcos motores do desenvolvimento, mas muito diferente do irmão.
Quando Caê completou um ano de vida, ele ainda não ficava em pé, nem mesmo com apoio. A família ligou um alerta. “Buscamos a ajuda de especialistas, para ter mais de uma opinião, foram duas ortopedistas e ambas solicitaram exames de imagem (ultrassom, ressonância magnética dos joelhos, da coluna cervical e cérebro). Como não havia lesões ou malformações encaminharam para a neurologista, que nos encaminhou para o geneticista”, conta Larissa.
Na primeira consulta, o médico geneticista suspeitou de AME (Atrofia Muscular Espinhal). O especialista coletou o material e em menos de 15 dias fechou o diagnóstico. O exame genético comprovou a suspeita inicial. “Apesar de ser uma notícia dura ela soou com algum grau de alívio, porque agora nós sabíamos o que estava acontecendo e podíamos buscar as melhores condutas”, revela a mãe de Caê.
Entre a investigação médica e o diagnóstico final, o bebê, além de não evoluir, começou a perder movimentos. Caê parou de engatinhar e apenas se arrastava. As mãos ficaram mais fracas e trêmulas. Ele já não tinha capacidade para erguer os braços e caia quando sentado. A fraqueza muscular era um sinal de que os neurônios dele estavam morrendo. “É muito triste ver seu filho definhando na frente de seus olhos e você não poder fazer nada além de buscar ajuda”, declarou Larissa.
Com a orientação médica, a família iniciou um tratamento com medicações que estão disponíveis pelo SUS: a Nusinersena e o Risdiplam. Enquanto a Nusinersena é uma injeção aplicada diretamente na coluna a cada 4 meses, o Risdiplam é um medicamento oral que deve ser tomado todos os dias, ambos são para o resto da vida. Caso não sejam administradas corretamente os sintomas podem avançar novamente.
A família teve que optar entre um dos dois medicamentos. Acabaram decidindo pelo Risdiplam porque era o que estava mais disponível. “Conseguimos na Farmácia do Componente Especializado (farmácia de alto custo) em Cuiabá em dois dias depois do diagnóstico. Consideramos esse tempo recorde e só foi possível porque outras 12 crianças já tomavam o medicamento e havia unidades disponíveis na farmácia”, explicou Larissa.
Com o Risdiplan, Caê sobrevive, mas é no Zolgensma que está a esperança de uma vida boa. Essa terapia genética, aplicada em uma única dose, é o que há de mais avançado para o tratamento de AME.
O Zolgensma obteve registro junto à Agência Nacional de Vigilância Sanitária (Anvisa), incorporada ao SUS em 2022 e incluído no Rol da Agência Nacional de Saúde Suplementar (ANS).
Caso recente em Mato Grosso
Em agosto de 2022, a Justiça Federal negou o Zolgensma para o pequeno Henry Erik, na época com um ano e 2 meses. O bebê residia com a família em Várzea Grande (MT). O caso repercutiu na imprensa, o Ministério Público recorreu da decisão da 21ª vara, mas a sentença não foi revertida.
No dia 12 de março desse ano, juiz federal da 4ª vara do Distrito Federal, Gustavo de Mendonça Gomes, concedeu uma antecipação de tutela, garantindo que uma criança com menos de 2 anos recebesse do SUS o Zolgensma.
Que doença é essa?
A AME é uma doença degenerativa e genética causada pela falta do gene SMN1 (Survival Motor Neuron 1), o que altera a produção da proteína SMN, essencial para a sobrevivência dos neurônios motores presentes na medula espinhal. A redução da proteína SMN leva à degeneração progressiva dos neurônios motores, que controlam os músculos voluntários.
A AME causa fraqueza muscular progressiva, atrofia muscular, hipotonia e perda de reflexos profundos. As manifestações clínicas variam de acordo com a quantidade de proteína SMN que a pessoa é capaz de produzir e é dividida em tipos de acordo com a gravidade. O tipo 1 e 2, por exemplo, são a forma mais grave e se manifesta nos primeiros meses de vida, com grande comprometimento da deglutição e da respiração. Enquanto os tipos 3 e 4 têm início mais tardio e sintomas mais leves.
O diagnóstico de AME é gratuito e em breve deve estar disponível no teste do pezinho, o primeiro exame que um recém-nascido é submetido. Quem tem AME não pode tomar uma série de medicamentos e corre risco de morte. “O diagnóstico muito mais que uma má notícia é uma bússola que nos aponta os novos caminhos”, ressalta a mãe do Caê.
Em sua experiência, Larissa recomenda ficar de olho nos sinais que precedem uma doença. O atraso no desenvolvimento, por exemplo, pode esconder algo mais grave. “Agradeço por termos tido essa percepção antes dos sinais mais graves aparecem e as vezes me pergunto se não podíamos ter identificado mais cedo ainda. Mas sabemos que não dá para voltar ao passado para aumentar o sinal de alerta. O que nos resta é alertar e apoiar as pessoas que assim como nós são pais atípicos. Para que, apesar da deficiência, nossos filhos possam ter vida plena”, ensina Larissa.
Notícias dos Poderes
Médico opera brasileiro a 12 mil km de distância e entra para o Guinness
Procedimento quebrou o recorde de telecirurgia robótica de maior distância já registrada no mundo; o cirurgião operou diretamente do Kuwait, enquanto o paciente estava em Curitiba
23 de Outubro de 2025 as 11h07SES confirma primeira intoxicação por metanol em MT e vítima está cega
22 de Outubro de 2025 as 20h58Hospital Regional de Sinop promove conscientização sobre câncer de mama para servidoras
Ação é resultado de parceria entre a Secretaria de Estado de Saúde e a Universidade Federal de Mato Grosso
22 de Outubro de 2025 as 14h30Prefeitura de Sinop leva vacinação e pesagem do Bolsa Família ao Residencial Nico Baracat neste sábado (25)
22 de Outubro de 2025 as 11h49SES promove captação de múltiplos órgãos no Hospital Regional de Sinop
Equipe de Cuiabá viajou para Sinop para realizar captação de dois rins e duas córneas, que vão salvar a vida de quatro pacientes
20 de Outubro de 2025 as 19h29Dia D de vacinação mobiliza unidades de Sinop e mais de 1,5 mil doses são aplicadas
20 de Outubro de 2025 as 14h01Pela primeira vez em 20 anos, número de fumantes cresce no Brasil
Proporção de adultos fumantes saltou de 9,3% para 11,6%
20 de Outubro de 2025 as 10h26AVC mata uma pessoa a cada seis minutos no Brasil
Custo hospitalar com internação foi de quase R$ 1 bi em seis anos
20 de Outubro de 2025 as 09h24